Day 1 :
Keynote Forum
Norris Stanley Nahman
Medical College of Georgia-Augusta University, USA
Keynote: The regulation of indoleamine 2, 3 dioxygenase and its role in a porcine model of acute kidney allograft rejection
Time : 10:00-10:45

Biography:
Stanley Nahman joined the Faculty at Ohio State University College of Medicine after completing his fellowship at the same institution in 1987. In 2004, he became Director of Nephrology division of University of Florida Jacksonville. In 2010, he joined Nephrology division at Medical College of Georgia, where he has directed the Department of Medicine Translational Research Program since 2013, leads a USRDS data mining group, and is PI of a basic research lab studying the tolerogenic properties of enzyme IDO. In 2017, he became Co-chair of American Society of Nephrology in-training exam question writing group.
Abstract:
In kidney transplantation acute rejection is the most common cause of late allograft loss. Changes in indoleamine 2, 3 dioxygenase (IDO) activity, which catabolizes the degradation of tryptophan to kynurenine, may predict rejection. However, when used therapeutically, IDO is immunosuppressive in rodent kidney transplantation. Thus, the increase in IDO activity observed in acute allograft rejection is insufficient to prevent rejection. To address this question, we assessed the regulation of IDO and its role in acute rejection in a porcine model of kidney transplant. In tissue samples form rejecting kidney allografts we showed a 13 fold increase in IDO gene transcription, and 20 fold increase in IDO enzyme activity when compared to autotransplanted kidneys. Allografts also demonstrated an over 4-fold increase in tissue IFN-ï§, with marked increases in TNF-ï¡, TNF-ï¢ and IL1-ï¢. Rejecting allografts also showed down regulation of kynurenine 3-monooxygenase (KMO) gene transcription and protein levels. KMO generates the immunosuppressive kynurenine 3-hydroxykynurenine (3-HK) from kynurenine. The results of these studies demonstrate a clear association between rejection and increased allograft IDO expression, likely driven in part by IFN-ï§ and facilitated by other cytokines of the allogeneic response. Moreover, the loss of downstream enzymatic activity in the IDO metabolic pathway may suggest novel mechanisms for the perpetuation of rejection in the early transplant period.
Keynote Forum
Xiaonan Wang
Emory University, USA
Keynote: Muscle-derived miR-26a mediate cardiac fibrosis through exosome in chronic kidney disease mice
Time : 10:45-11:30

Biography:
Dr. Xiaonan Wang gained his MD in 1982 from Peking Union Medical College, Beijing, China. She finished her post-doc training in 1991 in the University of Colorado HSC, Denver, CO and in 1997 in Emory University, Atlanta, GA, USA. Currently, Dr. Wang is an Assistant Professor of Renal Division, Department of medicine. She has published 50 papers in peer review journal. Since 1997, Dr. Wang has focused on investigation of the molecular/cellular mechanisms that lead to protein malnutrition in, diabetes, chronic kidney disease and aging in order to develop therapeutic strategies for treatment.
Dr. Wang uses transgenic mice, virus (adenovirus, adeno-associated virus (AAV) and lentivirus) mediated gene transfer and cell culture systems to test her hypotheses.
Abstract:
Uremic cardiomyopathy and muscle atrophy contribute to CKD-induced morbidity and mortality. Exosomes, natural carriers of many signal molecules including microRNA (miR), mediate organ-to-organ communication. We hypothesized that miR-26 would benefit both CKD-induced muscle wasting and cardiomyopathy through exosome-mediated muscle-heart crosstalk. We used an engineered exosome vector, which contains an exosomal membrane protein gene Lamp2b fused with muscle specific surface peptide for targeting delivery. Exosome encapsulated miR-26a precursor RNA (Exo/miR26) were injected into the tibialis anterior (TA) muscle of CKD mice (5/6 subtotal nephrectomy) for 10 weeks. miR-26a was decreased in skeletal muscle and heart of CKD mice. Uremic serum enhanced secretion of miR-26a exosomes in cultured C2C12 skeletal and H9C2 cardiac muscle cells. The intervention of Exo/miR26a increased the expression of miR-26a in skeletal muscle and heart, as well as increased muscle cross-section area and decreased CKD-induced up-regulation of atrogin-1 and MuRF1. Curiously, cardiac fibrosis lesion was partially depressed, and FoxO1, α-SMA, connect tissue growth factor (CTGF), fibronectin and collagen1α were decreased in CKD mice with intramuscular injection of Exo/miR-26a. Echocardiography showed that the percentage of ejection fraction was increased in CKD mice treated with Exo/miR26a. Using fluorescence dye labeled Exo/miR26a; we found that the fluorescence intensity in heart was correlated with skeletal muscle, examined by linear regression. We found that miR-26a directly inhibits FoxO1 and CTGF, which provided mechanism for inhibition of muscle atrophy and cardiac fibrosis by Exo/miR26a. Overexpression of miR-26a in muscle prevents CKD-induced muscle loss and attenuates cardiac fibrosis via exosome-mediated muscle-heart crosstalk.
Keynote Forum
Maria Hernandez-Fuentes
UCB Celltech, UK
Keynote: Biomarkers of tolerance in kidney transplantation: When predicting tolerance adjustment for confounding factors is imperative
Time : 11:50-12:30

Biography:
Maria Hernandez-Fuentes studied Medicine at Universidad Complutense and then completed PhD in Immunology at Universidad de Alcalá, both in Madrid, Spain. She then moved to the UK, Imperial College London, working on alloimmune responses. In 2005, the research group moved to King’s College London and since then she led the biomarker research group of the MRC Centre for Transplantation. She has a long standing interest in understanding and quantifying alloimmune responses and immune monitoring in kidney transplantation; particularly looking at obtaining evidence of tolerance.
Abstract:
We and others have previously described signatures of tolerance in kidney transplantation showing differential-expression of B-cell related genes and relative expansions of B-cell subsets. However, in all of these studies, the index groups namely the tolerant recipients were not receiving immunosuppressive (IS) treatment unlike the rest of the comparator groups. The work will demonstrate that the expression of the previously reported signature was biased by IS regimens, which also influenced transitional B-cells. We have defined and validated a new gene-expression signature that was independent of drug effects and also differentiated tolerant patients from healthy controls and have validated this signature in a number of cohorts. We will demonstrate how adjustment for IS-drug intake does not obliterate the contribution of genes to tolerance, when this exists; but it does indeed remove the effects ascribable to pharmacological immunosuppression and, thus, reveals underlying tolerance characteristics. Consequently, we would argue that IS regimens do affect the expression of many genes (although not all) and require adequate investigation. When IS are, indeed, altering the expression of signature genes, investigators should adjust for IS-drug intake. Only a similar approach will make the conduct of pilot clinical trials for IS-minimization safe, and hence allow critical improvements in kidney post-transplant management.
- Nephrology | Kidney Transplantation | Glomerular Disorders | Renal Pathology-Immunology | Kidney Diseases |
Location: Olimpica 3 + 4

Chair
Norris Stanley Nahman
Medical College of Georgia-Augusta University, USA

Co-Chair
Maria Hernandez-Fuentes
UCB Celltech, UK
Session Introduction
Zhen Su
Wenzhou Medical University, China
Title: Muscle-derived exosome/miR-29 attenuates kidney fibrosis in UUO mice

Biography:
Zhen Su has completed her MD from Wenzhou Medical University; PhD from Second Military Medical University, and; Post-doctoral studies from Emory University School of Medicine. She is the Vice-Principle Investigator of renal division in the First Affiliated Hospital of Wenzhou Medical University. She has published more than 40 papers in reputed journals and had oral presentation in ASN 2005 (American Society of Nephrology) meeting and WCN 2007 (ISN World Congress of Nephrology) meeting.
Abstract:
We hypothesized that intramuscular injection of exosome encapsulated miR-29 would counteract unilateral ureteral obstruction (UUO)-induced muscle wasting and renal fibrosis via exosome-mediated muscle-kidney crosstalk. Exosomes containing miR-29 (Exo/miR29) were prepared from the satellite cells and injected into the tibialis anterior muscle of UUO mice for one to four weeks. The expression of miR-29 was decreased in skeletal muscle and kidney of UUO mice. Serum from UUO mice enhanced secretion of exosome-encapsulated miR-29 from cultured C2C12 skeletal muscle and HEK293 renal cells. The intervention of Exo/miR29 increased muscle cross-section area and decreased UUO-induced upregulation of TRIM63/MuRF1 and FBXO32/atrogin-1. Curiously, BUN was decreased in the mice treated with Exo/miR29. In addition, renal fibrosis was partially depressed in the UUO mice with intramuscular injection of Exo/miR29. This was confirmed by decreased TGFβ, alpha-smooth muscle actin (aSMA), fibronectin, collagen 1A1 and 4A1 in the kidney of UUO mice by muscle-derived miR-29. When we used fluorescence-labeled Exo/miR-29 to trace the Exo/miR route in-vivo we found that fluorescence was significantly visible in both injected and un-injected muscle and in kidneys. The fluorescence intensity in kidney correlated with skeletal muscle. We found that miR-29 directly inhibits TGF-β3 in cultured kidney cells. We conclude that exosomes play a critical role in muscle-kidney crosstalk. Muscle-derived Exo/miR29 not only ameliorates skeletal metabolism, but also attenuates UUO-induced kidney fibrosis by down-regulating TGF-β pathway proteins and extracellular matrix components.
Banan Abbas Mustafa Osman
Bristol Urological Institute, UK
Title: Renal function preservation following high grade blunt renal trauma: A major trauma centre experience

Biography:
Banan Abbas Mustafa Osman is a British urology trainee joined University of Medical Science and Technology. She is an Honorary Clinical Tutor for the Severn School of Surgery. She is also a teaching faculty member at Royal College of Surgeons England. She was appointed annually over three years as a Surgical and Urology trainee representative in Junior’s Doctor’s Forum in the Royal Marsden NHS foundation Trust & North Bristol NHS Trust. She was a Clinical Investigator in VORTEX Clinical Trial. She held a position of Assistant Director of training in SMA International Training School.
Abstract:
Kidneys are the most common genitourinary organ injured following trauma. 90-95% of renal injures are secondary to blunt aetiology. Conservative management of renal trauma has brought the question of long term complications and renal function preservation. This study evaluates renal function preservation following high grade blunt renal trauma in a level 1 trauma centre in the United Kingdom between January 2012 and December 2015. Trauma Audit & Research Network (TARN) data base, patient’s electronic records and radiology scans were reviewed and results were analysed. 4611 patients with major trauma admitted into the hospital, of which 1.8% (83/4611) patients were identified with urological blunt trauma. The mean age of population was 44 years; range 12-90.5 years with a male predominance (82%). Renal injury counted for 51.8% of urological blunt trauma patients with (26) 45% grade I, (9) 15% grade II, (4) 7% grade III, (15) 26% grade VI, (4) 7% grade V. Conservative management was successful in patients with grade IV renal trauma with 6.7% morbidity with long-term hypertension. None of grade IV developed any renal function deterioration. Over all, long term follow up (four years average) of patients, revealed 95% of patients preserved their pre trauma renal function. This study reveals preserved renal function in over 95% of patients with blunt renal trauma. We therefore promote conservative approach of high grade renal injury with the objective of renal function preservation; however, certain situations still require intervention.
David Tovbin
Emek Medical Center, Israel
Title: Correcting acidosis during hemodialysis: Current limitations and potential solution

Biography:
David Tovbin has completed his MD at Hebrew University School of Medicine, Jerusalem, Israel and his Nephrology Fellowship at Southwestern Medical Center at Dallas, USA. He is currently Head of Nephrology at Emek Medical Center, Afula, Israel and Assistant Clinical Professor at Faculty of Health Sciences, Technion, Institute of Technology, Haifa, Israel for the last seven years. He serves as Head of Hemodialysis Forum of Israeli Society of Nephrology and Hypertension. His main research interests include “Acid-base status and anemia and iron deficiency correction in hemodialysis patients”.
Abstract:
The deleterious catabolic and pro-inflammatory effects of acidosis in hemodialysis (HD) patients and the importance of its correction for limiting mineral bone disorder (MBD), are well known. Although oral base therapy could be a solution for correcting acidosis in HD patients, it increases their already enormous medication load. Thus, this approach is not commonly used. Therefore, we need to rely more on correcting acidosis during the HD procedure. This is difficult to achieve because HD is an intermittent therapy that tries to correct in few hours processes that occurred in few days. In addition, most the acid load accumulates in the extensive extra-plasma compartments while the initial changes during HD are induced through the relatively restricted plasma compartment. Thus, the currently used fixed dialysate bicarbonate concentrations are associated with pre-HD acidosis and intra-dialytic alkalosis. Recently, large scale studies have demonstrated that using higher dialysate bicarbonate concentration and intradialytic alkalosis are deleterious. Decreasing dialysate bicarbonate concentration from an initially high concentration may be a means of correcting acidosis with limited intradialytic alkalosis. Caution may be required with changes in potassium and ionic calcium levels. Some evidence, as well as theoretical considerations, supports such an approach.
Manal Abd Elsalam
AL-Azhar University, Egypt
Title: Evaluation of sclerostin serum level and bone density status in children on regular haemodialysis

Biography:
Manal Abdelsalam completed her MBBCh and, then Residency of Pediatrics at AL-Azhar University, Cairo, Egypt. She is an Assistant Professor of Pediatrics at Al-Azhar University, Cairo, Egypt.
Abstract:
Bone disease is frequently observed in chronic kidney disease (CKD) and increases a patient’s risk for fracture. Sclerostin is an osteocyte-derived negative regulator of bone formation. Aim of this study is to assess sclerostin serum level as a bone marker in children with CKD on regular hemodialysis and detect the association between sclerostin serum level and bone density status. The study conducted on 25 children with CKD on regular HD and 25 age- and sex-matched healthy children as controls, complete blood picture, BUN, serum creatinine, parathyroid hormone, alkaline phosphatase, calcium, phosphorous and sclerostin serum level, also DEXA scan were measured in both groups. There was significant increase in sclerostin serum level in patients compared to controls with nine patients (36%) have low bone mineral density (BMD) with z score under -2.0, 8 of them have low BMD in both the neck of femur and lumber spines and one of the patients only have low BMD in the lumber spines. There is significant increase in sclerostin serum level in patients group with low BMD compared with patients with normal bone density. There is significant positive correlation between sclerostin serum level and (ALK phosphatase, PTH) while there is significant negative correlation and serum Ca. Sclerostin is 100% specific and sensitive as a marker of bone disease in children of regular hemodialysis. Elevated sclerostin levels are consistent with low bone density and appear to be independent predictor of reduced bone mineral density in CKD children on regular hemodialysis.
Han-Yu Tsai
Chang Gung Memorial Hospital, Taiwan
Title: Huge retroperitoneal epithelioid angiomyolipoma: A case report

Biography:
Han-Yu Tsai has completed his MD degree in medicine from Chang Gung University, Taiwan. He is currently a resident doctor in the division of urology, department of surgery, Chang Gung Memorial Hospital. He has published one case report and has been a speaker in the 2017 American Urological Association about researches in epithelioid angiomyolipoma.
Abstract:
Angiomyolipoma (AML) is a type of tumor in the perivascular epithelioid cell neoplasm (PEComa) family and is the most common benign solid renal neoplasm. Epithelioid angiomyolipoma (EAML), having malignant potential, is considered a rare variant of angiomyolipoma. The most common site of EAMLs is kidney, and extra-renal EAMLs are very uncommon. To our acknowledgment, only six cases of retroperitoneal EAML were reported in the English literature. Presented here is a 46-year-old female with a giant (20 cm×15 cmx15 cm) retroperitoneal epithelioid angiomyolipoma. On pre-OP CT, the tumor was encapsulated with soft tissue and displacing the left kidney upwards and compressing abdominal aorta. The cystic mass was decompressed for 1300 ml of blood and was dissected carefully from the left kidney. Pathology revealed tumor composed largely of polygonal epithelioid cells with abundant eosinophilic cytoplasm, marked nuclear pleomorphism and hyperchromasia, and occasional bizarre tumor giant cells. There were no identifiable renal tissues seen. Hence, the tumor is most likely arising from the renal capsule. Follow-up CT urography and creatinine after three years revealed no evidence of recurrence or metastasis.
Amir Abbas Farshid
Urmia University, Iran
Title: Ultrastructural alterations of renal tissue in a male patient with fabry disease

Biography:
Amir Abbas Farshid is a Professor of Veterinary Pathology, Faculty of Veterinary Medicine, as well as Head of Electron Microscope Center, Urmia University, Urmia, Iran, with more than 85 research papers published in reputed journals.
Abstract:
Fabry disease is an X-linked lipid storage disorder due to deficient lysosomal alpha galactosidase A. Kidney biopsy was done on a 19 year old male patient with complaint of acroparesthesia, maculopapular skin lesions and cornea verticillata. Kidney biopsy tissue was processed and examined by electron microscopy. Changes were inclusion bodies in the cytoplasm of the renal cells. These inclusions were osmophilic with concentric lamellation of clear and dark layers, showing onion skin appearance. The podocytes were mostly affected and some of the foot processes were fused. Cross-sections of collagen fibers were also evident, indicating fibrosis. The ultra-structure of the kidney clearly showed the intracytoplasmic glycosphingolipid accumulation in renal cells, responsible for progressive decline in renal function which could lead to kidney failure. The final diagnosis of Fabry disease was confirmed. In the present case-study, electron microscopy proved to be a valuable diagnostic aid.
- Chronic Kidney Disease | Kidney Cancer | Hypertensive Associated Kidney Diseases | Dialysis | Urinary Tract Infections |
Location: Olimpica 3 + 4

Chair
Vladimirs Strazdins
Medical Society Gailezers, Ltd., Latvia

Co-Chair
Nadica Ristoska-Bojkovska
KB Acibadem Sistina, Republic of Macedonia
Session Introduction
Banan Abbas Mustafa Osman
Bristol Urological Institute, UK
Title: Medical expulsive therapy for ureteric stones: Analysing the evidence from systematic reviews and meta-analysis of powered double-blinded randomised controlled trials

Biography:
Banan Abbas Mustafa Osman is a British urology trainee joined University of Medical Science and Technology. She is an Honorary Clinical Tutor for the Severn School of Surgery. She is also a teaching faculty member at Royal College of Surgeons England. She was appointed annually over three years as a Surgical and Urology trainee representative in Junior’s Doctor’s Forum in the Royal Marsden NHS Foundation Trust & North Bristol NHS Trust. She was a Clinical Investigator in VORTEX Clinical Trial. She held a position of Assistant Director of training in SMA International Training School.
Abstract:
Urinary tract stones affect 1–15% of the general population and the incidence is on the rise. Annual costs for stone disease have rapidly increased over the years and most patients with ureteral colic or other symptoms seek medical care. This has led to a plethora of research into the field aiming at finding a medication that will increase stone passage, shorten time to passage, and alleviate pain. The role of medical expulsive therapy (MET) is debatable with studies showing benefit for medical expulsive therapy (MET), whilst others reported no benefit. However, despite the multitude of trials published, the debate remains, as most of the research is riddled with bias and confounding factors. We therefore conducted a Cochrane style systematic review and a pooled meta-analysis on published literature from 1990 to 2016, to include low risk of bias (RoB) randomised controlled trials (RCTs) and a power calculation to investigate the efficacy and safety of medical expulsive therapy (MET). Pooled analysis of powered RCTs with low RoB would suggest, MET with the use of an a-blocker does increase stone expulsion rates (a-blockers 78% vs. 71% control) (P<0.001). Furthermore, their role is more significant for larger (>5 mm) stones (a-blockers 75% vs. 61% control) (P=0.02) and stones in the lower ureter (a-blockers 83% vs. 72% control) (P<0.001). However, MET was associated with side-effects, albeit not severe.
Vladimirs Strazdins
Medical Society Gailezers, Ltd., Latvia
Title: Recurrent urinary tract Infections in adults in Latvia: Additional findings from 2014 study

Biography:
Vladimirs Strazdins completed his Graduation from Riga Stradins University in 1977 and dedicated his further carrier to nephrology and pediatric nephrology. From 1992 till 2009, he worked as a Head of Nephrology at University Hospital for Children in Riga. From 2004 till 2012, he was a member of European Pediatric Dialysis working group, participating in creation of European guidelines on the subject. In 1999, he was awarded with Special Recognition Award for developing the pediatric ESRD treatment program in Latvia and Lithuania; in 2013 awarded with Honor Medal Tempus Hominis 2nd degree for outstanding achievements in medicine.
Abstract:
Antimicrobial resistance is a growing worldwide problem. Urinary tract infection (UTI) is not an exception. Furthermore, the recent trends may prohibit the further use of fluoroquinolones in UTI treatment. Therefore, it is crucial to regularly update the bacterial flora spectrum data and the efficacy of the recommended empiric treatment to make timely and appropriate amendments where necessary. This observational study was comprised in July-November 2014, when family physicians across Latvia submitted the anonymous patient data on recurrent UTI treatment in their practice. Bacterial flora spectrum in Latvian adult recurrent UTI population was fairly consistent with data from other European countries, with prevalent Escherichia coli cultures. The soluble nitrofuran derivate (NFD) - in Latvia Furamags ® in particular was clinically effective in all patients, even in culture-negative or NFD-resistant patients. There was not a single case without any improvement in controlled parameters – Furamags ® was clinically effective in all cases, including pyelonephritis. This confirms unique multifactorial antibacterial activities of NFDs, which simultaneously inhibit protein synthesis, aerobic energy metabolism, DNA synthesis, RNA synthesis and cell-wall synthesis, thus ensuring antibacterial activity even against the seemingly resistant flora. The current first-choice empiric treatment of recurrent UTI by NFDs may stay unchanged. Particular NFD used in Latvia (Furamags ®) is safe, well-tolerated and effective first-line UTI (including pyelonephritis) treatment choice.
Nadica Ristoska-Bojkovska
Clinical Hospital Acibadem-Sistina, Macedonia
Title: Congenital anomalies of the kidneys and urinary tract

Biography:
Nadica Ristoska-Bojkovska has completed Faculty of Medicine Specialist in Pediatrics; Subspecialist in Nephrology Faculty of Medicine in 1989 at Saints Cyril and Methodius University of Skopje, Skopje and she was employed at University Children's Hospital in Skopje 1990. She works at Clinical Hospital Acibadem-Sistina in Skopje from 2015. She started Pediatric specialization and passed the final specialization exam in 1996. She completed her Master Degree thesis in 2004 and; PhD in 2015 at Saints Cyril and Methodius University of Skopje, Skopje. She published many papers of scientific work and had fellowship at Charite Hospital, Berlin; Children's Hospital in Hanover and; Max-Dellbruck Center for Molecular Medicine in Berlin-Buch.
Abstract:
Congenital anomalies of the kidneys and urinary tract (CAKUT) are found in 3-6 out of 1000 of the newborns or according to some statistics they are represented in 0.5% of all pregnancies. Congenital abnormalities of the kidneys and urinary tract present a family of diseases of various anatomic spectrums, including renal anomalies, and anomalies of the bladder and urethra. This paper is motivated from the awareness that so far in Macedonia there has been no major or serious study prepared in relation to congenital kidney and urinary tract anomalies. The study was retrospective – prospective which means that it included newly diagnosed patients suffering from CAKUT, as well as those patients with already diagnosed and well defined CAKUT on the basis of imaging studies which have been processed according to the protocol for this study. A series of 749 pediatric patients with diagnosed congenital anomaly of the kidneys and urinary tract (CAKUT) has been analyzed at the Pediatric Clinic in the period from 2010 until 2015. In 25% CAKUT has been detected by prenatal ultrasound screening. The molecular diagnosis of patients with renal hypo dysplasia and renal agenesis can be achieved with copy number variation analysis in 10% of the patients. With this study we created a database of patients with syndromic and non-syndromic CAKUT, identified an unambiguous existence of the genetic factor through the familial ultrasound screening and existence of extra-renal abnormalities, thus enabling participation in future multicentric studies.
Hassan Kamal
Galilee Medical Center, Israel
Title: The impact of peritoneal glucose load on blood pressure in peritoneal dialysis patients

Biography:
Kamal Hassan is a specialist in Internal Medicine, Nephrology and Hypertension. He is Head of Peritoneal Dialysis Unit and In-patient Nephrology and Hypertension Department in the Galilee Medical Center, Naharyia, Israel. Also he is Clinical lecturer Faculty of Medicine. Main research interest: peritoneal dialysis and clinical nephrology.
Abstract:
Background: Hypertension is considered well-known independent risk factors for cardiovascular morbidity and mortality in peritoneal dialysis (PD) patients. Cardiovascular complications are the main cause of morbidity and mortality in patients with end-stage renal disease and dialysis patients. Peritoneal glucose load (PGL) contributes to the development of cumulative peritoneal membrane damage and increased permeability, leading to fluid accumulation and elevated blood pressure.
The effects of PGL on hydration status, systemic inflammation, left ventricular mass, depression and male sexual dysfunction among PD patients was evaluated in our peritoneal unit in several prospective cross sectional studies conducted in the last 5 years (2014-2018).The relationship between PGL and blood pressure was not investigated before. Based on the data obtained from the mentioned studies and additional data that received from the usual maintenance follow up visits we evaluated retrospectively the influence of PGL on blood pressure in patients on maintenance PD.
Methods: Office blood pressure measurements were used. If white coat hypertension is suspected, a 24 hour blood pressure study was performed to assess the patient's overall blood pressure profile using Mobil-O-Graph device for 24-hour ambulatory blood pressure monitoring (Manufacture: Industrielle Entwicklung Medizintechnik GmbH, D-52222 Stolberg, Germany). The hydration status was assessed by a whole-body bioimpedance technique (BIS) using a Fresenius Medical Care Body Composition Monitor (BCM) device (Fresenius Medical Care, Bad Homburg, Germany). The PGL was assessed by PGL index (PGLI), which refers to the net glucose content (monohydrated or unhydrated) (g) in the PD solutions administered in the daily PD prescription divided by the dry body weight (kg) assessed by BIS:
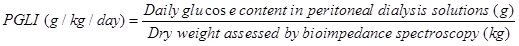
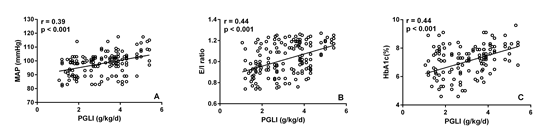
Results: One hundred fifty nine medical records of stable PD patients were evaluated retrospectively. Significant positive correlations were found between PGLI and mean arterial preasure (MAP), extracellular water (ECW)/ intracellular water (ICW) ratio and HbA1c. MAP, ECW/ ICW ratio and HbA1c were significantly higher in patients with PGLI > 3g/kg/day compared with those with PGLI ≤ 3g/kg/day.
Conclusions: PGL may be associated with higher blood pressure, overhydration and poor glycaemic control in PD patients. PGLI could be applied in managing PD patients as a practical tool for the quantitative assessment of the PGL. PGLI values bellow 3 g/kg/day should be targeted.
|
p |
Patients with PGLI > 3 g/kg/day n=76 |
Patients with PGLI > 3 g/kg/day n=83 |
|
|
<0.001 |
95.3±6.8 |
100.4±7.9 |
MAP (mmHg) |
|
<0.001 |
0.964±0.144 |
1.072±0.136 |
E/I ratio |
|
<0.001 |
6.56±1.13 |
7.52±0.94 |
HbA1c (%) |
Pai-Yen Pan
Chang Gung Memorial Hospital, Taiwan
Title: Mixed epithelial stromal tumor of the kidney: The male case and literature review

Biography:
Pai-Yen Pan has completed his MD degree at the age of 28 from Chang Gung University of Medicine. He is the resident doctor in the division of urology, department of surgery, Chang Gung Memorial Hospital. He has published a case report.
Abstract:
Mixed epithelial stromal tumor of the kidney (MESTK) is a rare genitourinary tract tumor. It was first presented by Michal and Syrucek in 1998.1 This tumor is characterized by its composition of both stromal solid areas and epithelial elements. Previous reports showed that MESTK attacks mostly middle-aged peri-menopausal women with estrogen therapy history, which indicates a correlation between MESTK and estrogen.2 However, rare cases were also reported in men and children.3 Even though malignant cases are rare, but they have also been reported for both genders. Since 2004, MESTK has been included in the World Health Organization renal tumor classification.
We report a 44-year-old Taiwanese male, with no history of hormonal therapy, who was found with a left renal tumor by self-health examination. Abdominal computed tomography showed an 11 x 15 cm enhanced heterogeneous soft tissue mass with calcification and minimal fatty content. He subsequently received radical left nephrectomy.
MESTK is a benign renal tumor with malignant potential. We should keep in mind that patients receiving hormonal therapy have a higher risk of developing cystic renal tumor, irrespective of their gender.
Manal M Thomas
National Research Centre, Egypt
Title: Genetic mutation in Egyptian children with steroid-resistant nephrotic syndrome

Biography:
Manal M Thomas is an Assistant Professor in Clinical Genetics department - Human Genetics and Genome Research division - National Research Centre, Cairo, Egypt. She has completed Master degree in Pediatrics from Medical School of Cairo University and PhD degree from Ain Shams University. She is a member of National Society of Human Genetics. She has many publications in medical genetics.
Abstract:
Nephrotic syndrome is the commonest etiology of proteinuria in children. Steroid-resistant nephrotic syndrome (SRNS) is defined by resistance to standard steroid therapy, and it continues to be one of the most intractable etiologies of renal failure. Molecular studies discovered specialized molecules in podocytes that play a role in proteinuria. Mutations in NPHS2 that encodes for podocin constitute a frequent cause of SRNS worldwide. This study aimed to screen for podocin mutations in SRNS Egyptian children and their parents. Our study included patients from 10 unrelated Egyptian families diagnosed with SRNS. Mutational analysis of the NPHS2 gene was performed by polymerase chain reaction amplification of the whole coding region of the gene and direct sequencing. Positive consanguinity was detected in five cases, and four of them had a positive family history of SRNS in a family member. Mutational analysis of NPHS2 revealed pathogenic mutations in four cases (40%) including a novel missense in one patient (c.1A>T; p.M1L). Our study concluded that mutations of NPHS2 gene are common among Egyptian children with SRNS. We support a model where ethnicity plays an important role in specific NPHS2 mutations, since a novel mutation was found in one patient in this study. Future study on a large number of Egyptian patients with SRNS is warranted to identify the actual genetic contribution of this gene in the development of SRNS in our population, which might help in patients’ prognosis and management.